Peptidbindung
Die AS werden durch Peptidbindungen aneinandergekettet. Polypeptide sind also kondensierte AS:
Bei der Kondensation von AS wird
Wasser abgespalten.
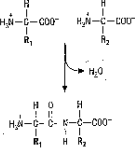
Die Eigenschaften der Peptidbindung liegen zwischen den Eigenschaften einer Imid- und einer Amidbindung.
Die Bildung der Polypeptide erfolgt mit Hilfe von aktivierten As an den Ribosomen der Zelle. Die Sequenz der AS bestimmt die Eigenschaften der gebildeten Proteine. Man unterscheidet am Protein
zwischen dem N-terminalen Ende und dem C-terminalen Ende (siehe Skizze).
Funktionelle Peptide spielen eine wichtige Rolle in Wissenschaft und Wirtschaft. Dabei sind die Anforderungen oftmals ebenso spezifisch wie die angeforderten Lösungen. Unternehmen wie die IPT mit ihrem Fachwissen
und langjähriger Erfahrung bieten dabei die erforderliche Zuverlässigkeit und Qualität. Mit einer Erfolgsquote von über 99% entwickelt JPT im Rahmen einer Custom Peptide Synthesis für jedes Peptid die passende Synthesemethode für ein hervorragendes Produkt.
Reinigung von Proteinen
Aussalzen
Beim Aussalzen von Proteinen beruht die Trennung auf der unterschiedlichen Löslichkeit der Proteine. Allerdings werden die Proteine bei dieser Methode durch Änderung des pH-Milieus,
Beigabe von Salzen oder Alkohol in ihrer Löslichkeit beeinflusst: sie fallen aus.
Mit dieser Methode lassen sich keine reinen Substanzen gewinnen, doch für eine grobe Einteilung der Proteine ist sie gut. Angewendet wird sie z.B. bei der Reinigung von Blutplasma (Fraktion
von Cohn).
Gelfiltration
Trennung der Proteine aufgrund unterschiedlichen Molekulargewichts (MG) bzw. Grösse. Die Gelfiltration ist eine Chromatographiemethode, die in einer mit porösem Material gefüllten
Säule durchgeführt wird.
Proteine mit kleinem MG dringen in die Poren ein und werden zurückgehalten, während Proteine mit grossem MG nicht in die Poren passen und die Säule somit schnell passieren.
Am Ende der Säule wird die Proteinlösung in verschiedenen Reagenzgläsern aufgefangen (à Fraktionen). Wegen der unterschiedlichen Durchlaufgeschwindigkeit befinden sich
in den ersten Reagenzgläsern die Proteine mit dem grössten MG und in den hinteren die mit dem kleineren MG. Es erfolgt somit eine grobe Trennung nach der Proteingrösse.
Die Säulen werden vor Gebrauch mit einer Standardlösung von Proteinen geeicht, von denen man die Grösse kennt und somit ihre Laufgeschwindigkeit stoppen kann Þ Vergleich
der Zeiten mit den zu untersuchenden Proteinen.
Ionenaustauscher
Der Ionentauscher nützt die unterschiedlichen Ladungen der Proteine zur Trennung aus. Beim Ionentauscher ist die Chromatographiesäule nicht mit porösem, sondern mit negativ
oder positiv geladenem Material gefüllt. Bsp: DEAE +
Das geladene Material wird mit einer Pufferlösung neutralisiert. Die zu trennenden Proteine ersetzen die Ionen des Puffers und binden an ihrer Stelle an das geladene Material und bleiben
somit in der Säule hängen. Anschliessend wird die Säule gewaschen und somit überschüssige Pufferionen entfernt. Um die Proteine aus der Säule zu gewinnen, wird
wieder Pufferlösung beigefügt. Nun verdrängen die Pufferionen die Proteine. Die Proteine verlassen die Säule. Alle Proteine der Lösung haben die gleiche Ladung: positiv
oder negativ, je nach Füllmaterial der Säule.
Neutrale Proteine werden im Ionentauscher nicht zurückgehalten!
Da jedes Protein seine eigene, vom pH abhängige Titrationskurve besitzt, kann durch die Wahl der Pufferlösung die Ladung der Proteine beeinflusst werden. Liegt ein Protein z.B.
bei pH 4 als Kation vor, ist es bei pH 6 vielleicht neutral.
Durch die Ionenaustauschchromatographie erhält man bis zu 80% reine Proteinlösungen!
Affinitätschromatographie
Die Affinitätschromatographie macht sich die unterschiedlichen physiologischen Eigenschaf-ten der Proteine zu Nutze.
An das poröse Material der Chromatographiesäule wird ein Ligand oder Antikörper gebunden (immobilisierter Ligand). Aus der beigefügten Proteinlösung bindet nur ein
einziges Protein an den Liganden/AK, d.h. der Ligand/AK ist spezifisch für ein Protein. Die Bindung Protein-Ligand/AK kann auf drei Arten wieder gelöst werden: Beigabe einer Pufferlösung,
Liganden im Überschuss oder durch kompetitive Substanzen.
Diese Methode ist sehr effizient, da oft ein einziges Protein aus der Lösung extrahiert werden kann.
Ultrazentrifugation
Bei der Ultrazentrifugation werden die Proteine nach ihrer Dichte, Form, Durchmesser und Masse getrennt.
Proteine haben unterschiedliche Sedimentationsgeschwindigkeiten, die abhängig ist von der Winkelgeschwindigkeit der Zentrifuge und von der Zentrifugationszeit.
Bei der Leerung des zentrifugierten Röhrchens erhält man verschiedene Fraktionen.
Diese Art der Proteinreinigung wird am Ende einer Reinigungsserie durchgeführt.
Native Elektrophorese
Diese Trennungsmethode macht sich die unterschiedlichen Ladungen (aber auch die Masse und Form) der Proteine zu Nutzen. Die Trennung erfolgt in einem elektrischen Feld, in wel-chen die Proteine
je nach Ladung wandern. Die Wanderungsgeschwindigkeit ist proportional zu ihrer Ladung, d.h. stark geladene Proteine wandern schneller.
Positiv geladene Proteine wandern zur Kathode
Negativ geladene Proteine wandern zu Anode
Die Proteine werden auf ein Gel aufgetragen, dann wird eine Pufferlösung, die die Proteine nicht denaturiert, beigefügt. Die Proteine werde so nach ihrer Ladung sortiert.
Denaturierende Elektrophorese
Die Trennung erfolgt nach Molekulargewicht. Die Proteine werden mit sodium dodecyl sulfat (SDS) denaturiert. Es bindet an alle Proteine und umgibt sie mit einem negativen Mantel, d.h. alle
Proteine sind nachher negativ geladen. Die Ladung ist proportional zur Grösse des Proteins, da grosse Proteine mehr SDS binden können als kleine.
Die so denaturierten Proteine werden wie bei der nativen Elektrophorese auf ein Gel aufgetragen. Da alle Proteine jetzt negativ geladen sind, wandern alle zur Anode. Ihre Wanderungsgeschwindigkeit
ist proportional zur Ladung und somit zur Grösse.
Diese Methode dient zur Bestimmung des Molekulargewichtes. Zum Vergleich werden parallel immer noch Proteine mit bekanntem Molekulargewicht, sogenannte Markerproteine, auf das Gel aufgetragen.
Mit dieser Methode werden sehr reine, doch denaturierte Proteine gewonnen.
Western Blot
Mit dem Western Blot können Proteine identifiziert werden. Es werden spezifische Antikör-per für ein Protein zur Identifikation benötigt.
Die Proteinlösung wird zuerst auf ein SDS-Polyacrylamid-Gel aufgetragen Þ Elektrophorese. Transfer der Proteinbanden auf ein Zellulose-Filter (= Blotting).
Nun wird ein spezifischer Antikörper zugegeben. Der Überschuss wird abgewaschen. Die Lage des gesuchten Proteins kann nun bestimmt werden. Dazu wird ein zweiter, radioaktiv markierter
oder ein an ein Enzym gekoppelter AK zugegeben, welcher spezifisch für den ersten AK ist. Radioaktive Markierung können auf einem Photofilm nachgewiesen werden. So lässt sich
die Bande auf der Membran erkennen.
Diese Methode liefert sehr gute Resultate, da AK meist nur an 1 Protein binden.
Isoelektrische Fokussierung
Die Isoelektrische Fokussierung trennt die Proteine aufgrund ihres isoelelektrischen Punktes (pI). Diese Methode ist sehr sensitiv, denn jedes Protein liegt bei einem genau bestimmbaren
pH neutral vor, d.h. die Totalladung ist gleich null.
Vorgehen: Die Proteine werden auf ein Gel mit einem pH-Gefälle aufgetragen. Die Proteine wandern im el. Feld bis zu dem pH-Wert bei welchem sie als neutrale Moleküle vorliegen,
d.h. bis zu ihrem isoelektrischen Punkt. Ein Proteine, das keine Nettoladung mehr trägt, wird in einem elektrischen Feld nicht mehr wandern. Die Proteine reagieren sehr sensibel, d.h.
auf pH Unterschiede von 0,1- 0,2.
Elektrophorese in zwei Dimensionen
Zuerst werden die Proteine auf einem Rundgel nach ihrem isoelektrischen Punkt getrennt. Daraufhin wird das Rundgel auf ein SDS-Gel gesetzt. Nun werden die Proteine noch nach ihrer Grösse
getrennt, sie wandern vertikal.
Diese Methode wird für Blutplasma verwendet. Sie dient zur Erkennung der vorhanden Mengen der Proteine. Ein pathogenes Blutplasma zeigt eine andere Verteilung oder andere Mengen and
Proteine. Einzelne Proteine können nun auch noch aus dem Gel geschnitten und analysiert werden (z.B mittels Massenspektrometrie).
Pathogene Hämoglobinformen lassen sich anhand der „fingerprints“, d.h. der verschiedenen Flecken auf dem Gel erkennen. Durch Analyse der abweichenden Flecken lässt
sich die veränderte Sequenz der AS bestimmen.
Durch die Analyse des Hämoglobins lässt sich aber auch die Phylogenese zurückverfolgen. Je unterschiedlicher das Hämoglobin ist, umso früher haben sich die Arten
getrennt. Die Veränderung der AS-Sequenz beruht auf Mutationen.
Proteinstrukturen
Analysemethode: Röntgenstrahlen werden auf einen Proteinkristall gestrahlt. Der Kristall bricht die Strahlen. Auf dem Photofilm hinter dem Kristall sind anhand des Diffraktions-bildes
die Konturen oder sogar die Atome des Proteins bestimmbar.
Nach der Reinigung werden die Strukturen der Proteine untersucht. Man unterscheidet 4 ver-schiedene Strukturen: die Primäre, die Sekundäre, die Tertiäre und die Quarternäre.
Primärstruktur: lineare Abfolge der AS
Sekundärstruktur: Interaktionen innerhalb der AS-Kette: Helices oder Faltblatt
Tertiärstruktur: Interaktionen zwischen den Sekundärstrukturen, d.h. räumliche Anord-nung des Proteins
Quarternärstruktur: Zusammensetzung aus Untereinheiten
Primärstrukur:
Bevor ein Protein überhaupt sequenziert werden kann, muss es in reiner Form vorliegen. Die Reinigung erfolgt mit oben beschriebenen Methoden. Eine andere Möglichkeit der Molekularbiologie
ist es, das Gen, welches für ein bestimmtes Protein kodiert, zu isolieren und das Protein so zu synthetisieren.
Da es zu schwierig ist, die Primärstruktur eines ganzen Proteins zu bestimmen, wird das Protein in Fragmente zerlegt. Die Zerlegung erfolgt mit Hilfe von chemischen Substanzen oder
Enzymen, die das Protein gezielt nach einer AS „zerschneiden“. Beispiele dafür sind CN-Br, welches die AS Met erkennt und Trypsin, welches nach Lys trennt.
Anschliessend werden die einzelnen Fragmente analysiert. Eine Methode zur Analyse ist die Edman-Sequenzierung: Bei der Edman-Sequenzierung bindet jeweils PITC ( Phenylisothio-cyanat) an
die N-terminale-AS. Mit Hilfe einer sauren Hydrolase wird das N-terminale Ende und PITC abgespalten. Diese N-termiale-AS kann nun in einer Dünnschichtchromatographie bestimmt werden.
Das Ganze wird mit dem Rest des Fragmentes wiederholt. Allerdings können so nur Fragmente mit bis zu 50 AS sequenziert werden, da Sekundärprodukte entstehen, die eine weitere Sequenzierung
behindern.
Die Bestimmung der N-terminale-AS kann auch zur Überprüfung der Reinheit eines Proteingemisches hinzugezogen werden: gleiche Proteine haben gleiche Enden.
Bei der Zerlegung in Fragmente und der anschliessenden Analyse der Primärstruktur geht allerdings die Reihenfolge der Fragmente verloren. Eine Rekombination ist dank der unter-schiedlichen „Angriffsaminosäuren“ der
verschiedenen zerlegenden Agenzien möglich. In grossen Teilen korrespondierende AS-Sequenzen können untereinander angeordnet werden und durch Überlappung kann die Reihenfolge
rekombiniert werden Sekundärstruktur
Die Peptidbindung ist eine planare, relative steife Verbindung mit einer Bindungslänge, die zwischen einer Amid und einer Imin-Bindung liegt. Der Winkel zwischen den einzelnen AS bestimmt über
die Art der Interaktionen.
Helix:
α-Helix:
- 3,6 AS pro Windung
- Stabilisierung durch Wasserstoffbrücken zwischen H und O, die 13 Atome voneinan-der entfernt liegen
- Stabilste Helix
π-Helix:
- die Atome der H-Brücken liegen 16 Atome auseinander.
- 4,3 AS pro Windung
Es gibt noch weitere Helices, die von nicht so grosser Bedeutung sind wie die genannten.
Faltblatt:
Stabilisierung parallel liegender AS-Ketten eines Proteins durch H-Brücken. Es gibt zwei Ar-ten von Faltblatt: antiparallel oder parallelen AS-Ketten
Antiparallele Faltblätter sind stabiler, da ihre H-Brücken gerade liegen. Tertiärstruktur
Die „Reste“ der AS helfen auch die Struktur zu stabilisieren. Es gibt verschiedene Arten der Interaktionen:
- Ionische Anziehung oder Abstossung zwischen COO- und NH3 verschiedener AS der Ketten, 20% der Stabilität
- H-Brücken zwischen OH und COO-
- Kovalente Interaktionen: Disulfidbrücken zwischen zwei Cysteinen
- Hydrophobe Interaktionen zwischen z.B. Isoleucin und Leucin, was eine Aggregation der hydrophoben „Reste“ bewirkt
Die Tertiärstruktur lässt sich verändern = Denaturierung der Proteine. Denaturierungen durch:
- Temperaturerhöhung
- pH-Variation
- Detergenzien
- Chaotropische Salze (z.B. Guanidinium-salz), d.h. denaturierenden Salze
- Harnstoff
Eine Renaturierung ist nach Entfernen der Denaturierungsagenzien z.T. möglich. Proteine liegen normalerweise auf dem tiefstmögliche Energieniveau.
Eine häufig auftretende Tertiärstruktur ist eine Abfolge von Helix- Schleife-Helix. Eine weitere ist die Fass-Struktur bei der mehrere Faltblätter parallel im Kreis angeordnet
sind.
Kalzium bindet oft in einer Schleife, die zwischen zwei senkrecht stehenden Helices liegt Þ bestimmt die Struktur von Ca2+ bindenden Proteinen (à sog. EF-hands). Bsp. Für
solche Calcium-bindende Proteine: Calretikulin, Calmodulin, … Quarternärstruktur
Viele Proteine liegen in Form von Polymeren vor. Es gibt Polymere aus gleichen Untereinheiten oder aus verschiedenen.
Beispiele:
Ferritin ( lagert Fe3+) , besteht aus 24 Untereinheiten mit je 4 a-Helices. Die Untereinheiten sind zu einem hohlen Ball angeordnet in dessen Mitte Eisen gelagert wird.
Hämoglobin: 2 a- Untereinheiten und 2 b-Untereinheiten. Hämoglobin ist nur in der Tetramerform aktiv. Jede Untereinheit enthält eine Porphyringruppe, welche ein Eisen(II)-Atom
enthält.
Auch bestimmte Enzyme sind nur in Polymerform aktiv: Beispiel: Lactatadehydrogenase (LDH), es besteht auch aus 4 Untereinheiten. Es gibt eine M und eine H Untereinheit. Je nach Gewebe ist
das LDH unterschiedlich zusammengesetzt. Mehr darüber unter den Enzymen.
Die 5 Formen des LDH lassen sich durch Elektrophorese des Blutes unterscheiden.
Pathologie: Nach einem Herzinfakt sind mehr Herzzellen zerstört, darum gibt es mehr H4- LDH im Blut als normal. Nach 6-8 Tagen hat sich der LDH-Spiegel wieder gesenkt.
Kollagen
Kollagen besteht aus 1052 AS, in einer repetitiven Sequenz angeordnet : (Gly-X- Y)337
33% aller AS des Kollagens sind somit Glycin, weitere 20-25 % bestehen aus den AS Prolin und Hydroxyprolin.
Man unterscheidet mehrere Arten von Kollagentypen:
I 90 % des adulten Kollagens, zu finden in Knochen, Sehnen...
II in Chondrozyten
III in Blutgefässen
IV in Basalmembran, Gefässen
V in Plazenta, Epidermis, Basalmembran
Kollagensynthese:
- Proteinsynthese, d.h. Translation an den Ribosomen
- Enzyme (Hydrolasen) verändern einige Proline und Lysine
- Bildung von Disulfidbrücken am C-terminalen Ende des Proteines und Formung einer Tripelhelix aus drei Kollagenketten
- Entfernen der Ende Þ Kollagenmonomer
- Polymerisation von Kollagenmonomeren, Glykosilierung (Glykosilierungsgrad ab-hängig vom Kollagentyp)
- Mineralisierung des Kollagens je nach Gewebe (Bsp: Knochen!)
Prolin verhindert die Ausbildung von a- Helices. Für die Umwandlung von Prolin in Hydroxyprolin ist Vitamin C nötig.
N.B : Proteine, die so reich an kleinen AS (Gly, Ala, Val,...) sind, werden imDuodenum durch die Protease Elastase abgebaut.
Hämoglobin (Hb)
Hb besteht aus vier Untereinheiten mit je einem Tetrapyrrolkern mit Fe2+, welches Sauerstoff binden kann. Ein Hb Molekül kann somit vier Sauerstoffmoleküle (O2) binden.
Eine Untereinheit ist aus 8 a-Helices aufgebaut (A-H). Der Kern befindet sich in einer Tasche des Proteins.
Hb ist nur als Tetramer aktiv: 2 a- und 2 b- Untereinheiten
1 α + 1 β = 1 Dimer
2 Dimere = 1 Tetramer
Durch die Aufnahme eines O2- Moleküls ändert sich die Konformation der Untereinheit. Die anderen Untereinheiten können nachher leichter Sauerstoff binden. Diese Art der Zusam-menarbeit
nennt man (positive) Kooperativität. Dementsprechend ist auch die Sättigungs-kurve des Hb sigmoidal, d.h. S-förmig (zuerst langsame Sättigung, dann schnellere).
Myoglobin, ein Molekül des Muskels, welches O2 speichert, kennt die Kooperativität nicht, da es nur als Monomer vorkommt. Seine Sättigungskurve verläuft linear.
Hb liegt in zwei Formen vor: als Desoxy- Hb und als Oxy- Hb. Desoxy- Hb hat seinen isoel. Punkt bei pH 6,6 und Oxy- Hb bei pH 6,8. Der unterschiedliche pH Wert im Blut, im arteriellen ist
er höher als im venösen, begünstigt die O2 – Abgabe ins Gewebe.
Sie unterscheiden sich aber auch in ihrem Spektrum. Desoxy- Hb hat sein Absorptions-maximum bei 560 nm. Oxy- Hb hat zwei Maxima bei 540 nm und 580 nm.
Lunge:
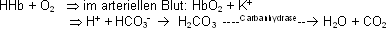
Gewebe:

Der Sauerstoff wird an das Gewebe abgegeben und Hb bindet H+ im venösen Blut und fliesst zurück zur Lunge
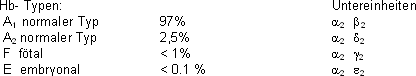
Die Untereinheiten unterscheiden sich minimal in ihrer Struktur.
HbF hat eine grössere Affinität für Sauerstoff. Dies ist eine wichtige Eigenschaft für die Sauerstoffversorgung des Fötus durch das Blut der Mutter. Das Hb des Fötus „entreisst“ quasi
dem Hb der Mutter den Sauerstoff ! Pathologische Hb Typen:
Bei pathologischen Hb Typen ist of nur eine As durch eine andere ersetzt!
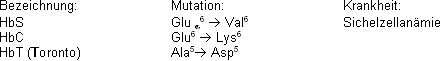
|
